BLAST是局部相似性基本查詢工具(Basic Local Alignment Search Tool)的縮寫。
是由美國國立生物技術信息中心(NCBI)開發的一個基於序列相似性的數據庫搜索程序,它能夠對生物不同蛋白質的氨基酸序列或不同基因的DNA序列與公開數據庫所有序列進行匹配比對,並從相應數據庫中找到相同或相似的序列。
BLAST可用於推斷序列之間的功能和進化關係,以及幫助鑑定基因家族的成員。BLAST還能發現具有缺口的能比對上的序列。BLAST可處理任何數量的序列,BLAST軟件提供了在線的比對工具,它不僅可以進行核酸序列之間的比對,也可以進行蛋白質與蛋白質以及蛋白質與核酸之間的比對。
通常根據查詢序列的類型(蛋白或核酸)來決定選用何種BLAST。
BLAST適用於本地查詢。可以下載公共數據庫,對於該數據庫的更新和維護是必不可少的。如果要直接到網上查詢也可以(即NET BLAST),但如果自己的序列很有價值的話,還是謹慎為宜。
例如我要對比某一段核酸序列,那就是點nucleotide blast
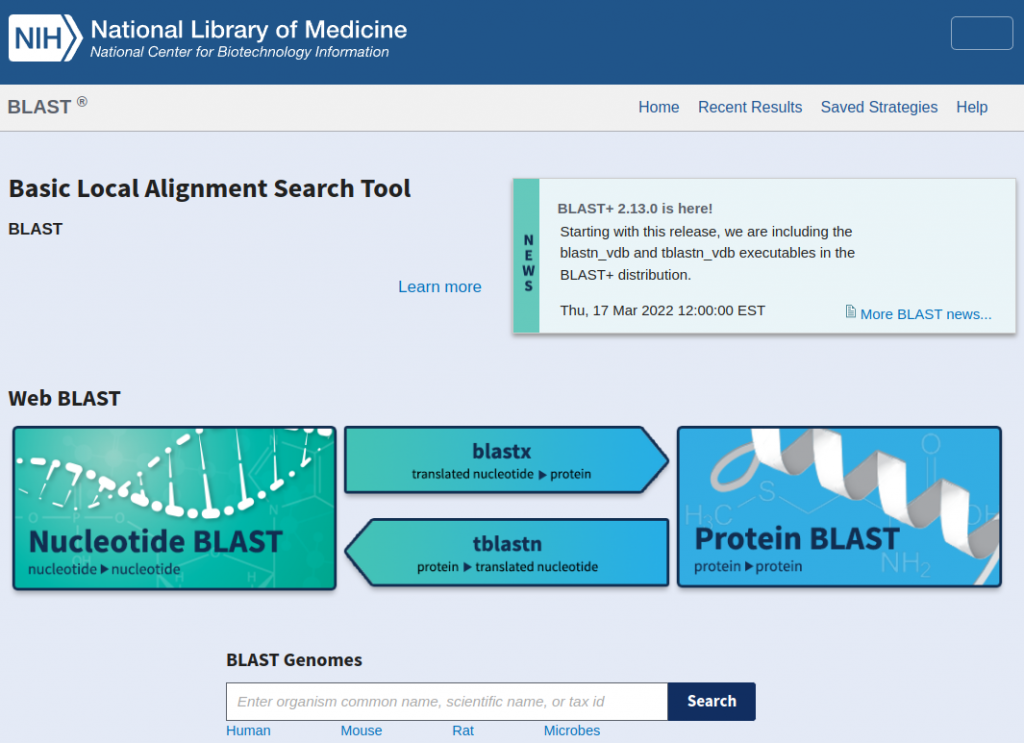
輸入序列以及一些想要的條件就可以查詢,同時也可以換別的blast比對工具。
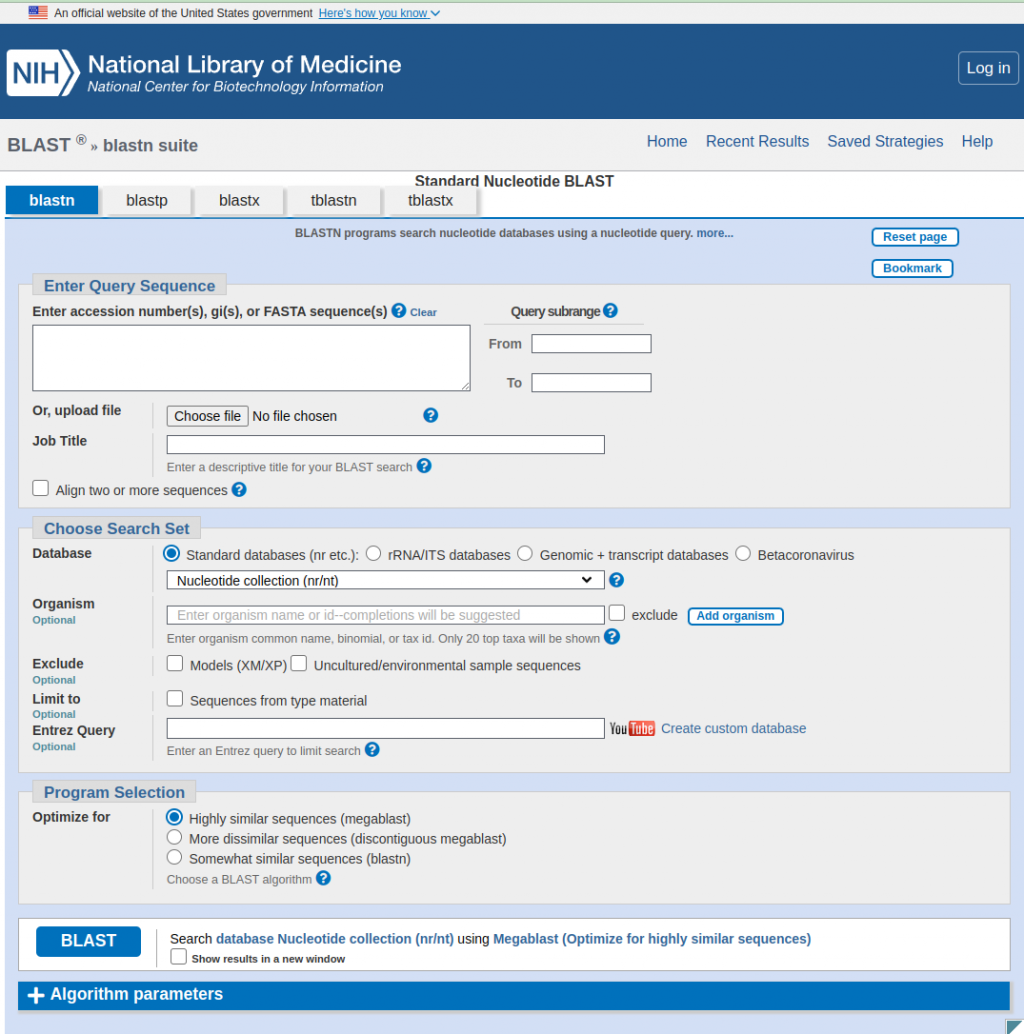
評價一個blast結果的標準主要有三項,E值(Expect),一致性(Identities),缺失或插入(Gaps)。加上長度(length)的話,就有四個標準了。
Score:序列比對過程中計算的得分值,得分越高,序列匹配結果越好。
Expect:表示隨機匹配的可能性。E值越小,序列越相似,E值越大,隨機匹配的可能性也越大。E值接近零或為零時,具本上就是完全匹配了。
Identities:序列相似性,匹配上的鹼基數佔總序列長的百分數。
Gaps:插入或缺失。用"—"來表示。
數據庫的下載:
ftp://ftp.ncbi.nlm.nih.gov/blast/db/FASTA/
conda create --name <myenv>
#activate your own environment
conda activate <myenv>
# BLAST installation (download latest BLAST version)
conda install -c bioconda blast
下載想要的ref genome DB,可以到NCBI資料庫找找要哪個物種,哪一版
例如我的:
建立自己的db
makeblastdb -dbtype nucl -parse_seqids -in GRCh38_latest_rna.fna -out human
-out human ,出來的db名字就都是human開頭,會跑出一對文件
-dbtype string,: string在(guess, nucl, prot)中選擇一個
裏面human開頭的都是建出來的DB
我的input sample : BLAST_sample_file.fa
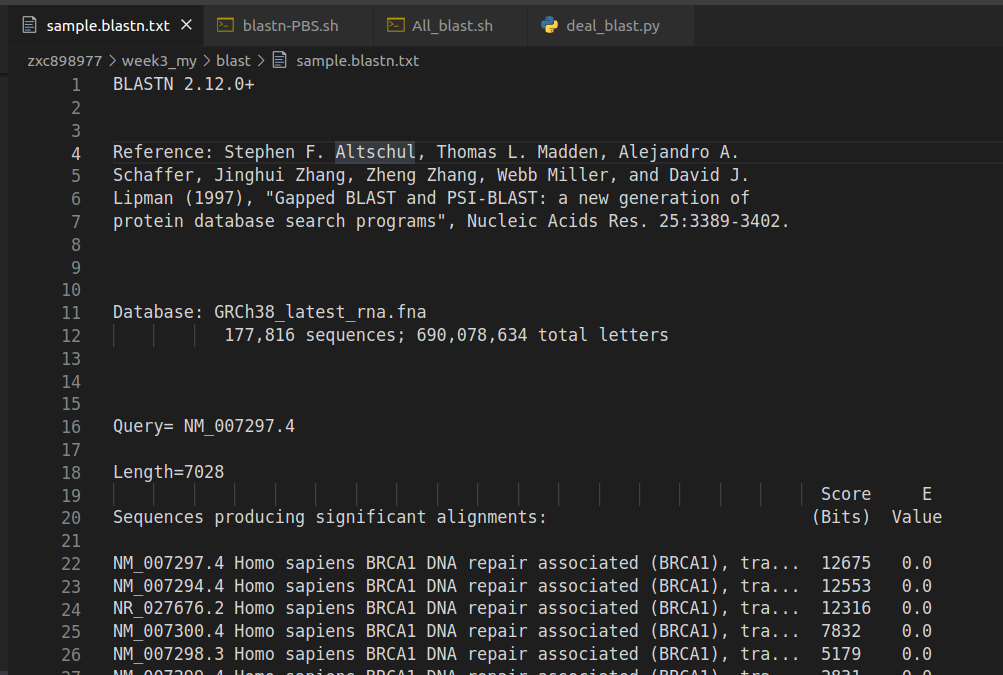
blastn -task blastn -query BLAST_sample_file.fa -db human -evalue 0.05 -out sample.blastn.txt
輸出來的sample.blastn.txt
Customized output table
blastn -task blastn -query BLAST_sample_file.fa -db human -outfmt "7 qseqid sseqid pident length mismatch gapopen qstart qend sstart send evalue bitscore staxids" -out sample.blastn.custom.txt
參數說明:
-query 需要檢索的序列文件名
-db 需要在哪個數據庫中檢索
-out 輸出文件名
-outfmt 輸出格式,一般選7即可,有0-18可選
-evalue E值閾值,高於這個E值的序列不輸出到結果中,默認值為10,建議設到10^-5以下
除此之外,還可以加入num_thread等參數,表示同時使用多少個線程來計算。其他更多參數可以使用
-help來查看
對應的header是:query id:查詢序列ID標識subject id:比對上的目標序列ID標識identity:序列比對的一致性百分比alignment length:符合比對的比對區域的長度mismatches:比對區域的錯配數gap openings:比對區域的gap數目query start:比對區域在查詢序列(Query id)上的起始位點query end:比對區域在查詢序列(Query id)上的終止位點subject start:比對區域在目標序列(Subject id)上的起始位點subject end:比對區域在目標序列(Subject id)上的終止位點e-value:表明在隨機的情況下,其它序列與目標序列相似度要大於S值的可能性,e-value的分值越低越好bit score:比對結果的bit score值
blastn -task blastn -query BLAST_sample_file.fa -db human -evalue 0.05 -out sample.blastn.txt
#BLAST Database error: Error: Not a valid version 4 database.
conda install -c bioconda blast=2.12.0
進階:megablast


 iThome鐵人賽
iThome鐵人賽